
With the advent of higher pressure boilers and more restrictive feed water requirements of turbine manufacturers, the ion exchange equipment manufacturers were called on to meet the challenge of purifying very large quantities of water which contained very small quantities of both dissolved and suspended contaminants. The major dissolved contaminants were sodium, chloride, and silica, while the suspended contaminants were oxides of iron and copper.
Conventional mixed bed demineralizers consisted of in-place regenerated resins. The anion resin was regenerated with caustic and the cation resin with acid. While the new condensate polishers were similar in nature to the conventional mixed bed demineralizers, the high operating pressure and flow rate required changes. The polishers were externally regenerated to limit the cost of duplicating the regenerant headers, valves, and piping. With external regeneration facilities, the freshly regenerated resins could be rinsed down to quality without risking contamination of the high-pressure boilers with traces of regenerant acid or caustic.
Initially, most of the power plant piping contained substantial quantities of copper in the condenser tubing. In order to reduce corrosion of the systems, ammonia was added to raise the pH. However, in the process of providing corrosion protection the ammonia dissolved some of the copper piping, and therefore industry decided to use all iron piping. Ammonia was added to raise the pH to between 8.8 and 9.6. The presence of ammonia in the condensate feeding the ion exchange polishers created a higher load on the hydrogen-form cation resin. Run lengths grew shorter as the ammonia concentration in the feed water was increased higher and higher to reduce iron transport in the system. The relatively short length of run required more and more regenerant, and problems were encountered in performing the number of regenerations required to keep the units on line. The industry then studied its early attempts at operating the polishers beyond the ammonia break. However, as the ammonia started leaking into the effluent, the sodium started to increase.
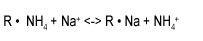 Let us examine the sodium equilibrium reaction with ammonia. We see that sodium in the influent to a polisher in the ammonia form produces ammonia in the effluent, with sodium being removed on the resin. The rate constant for this reaction is 0.75.
Let us examine the sodium equilibrium reaction with ammonia. We see that sodium in the influent to a polisher in the ammonia form produces ammonia in the effluent, with sodium being removed on the resin. The rate constant for this reaction is 0.75.
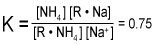 Assuming that the only cations present are sodium and ammonium, the sum of the sodium on the resin and the ammonium on the resin would be 100%
Assuming that the only cations present are sodium and ammonium, the sum of the sodium on the resin and the ammonium on the resin would be 100%
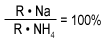 Rearranging the equation:
Rearranging the equation:
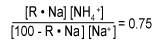 We see that sodium leakage in the effluent at equilibrium is a function of both the sodium on the resin and ammonia in the feed.
We see that sodium leakage in the effluent at equilibrium is a function of both the sodium on the resin and ammonia in the feed.
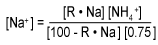
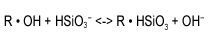 The equilibrium constant K,
The equilibrium constant K,  has been assigned an approximate value of 5 by Venderbosch (4).
Rearranging the equation as we have done with the cation resin, we see that the bisilicate ion concentration is a function of the hydroxyl ion concentration and the amount of bisilicate or silica on the resin.
has been assigned an approximate value of 5 by Venderbosch (4).
Rearranging the equation as we have done with the cation resin, we see that the bisilicate ion concentration is a function of the hydroxyl ion concentration and the amount of bisilicate or silica on the resin.
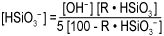 We can now see that:
We can now see that:
- As sodium on the resin increases, so does the sodium in the effluent (at constant ammonia concentration).
- As the ammonium concentration is increased (pH of the condensate), the sodium in the effluent will increase.
- As the ammonium concentration is increased, less sodium can be tolerated on the resin
- Ammonium hydroxide treatment of anion resin.
- Calcium hydroxide treatment of anion resin.
- Floating anion resin with sodium hydroxide.
- At any given pH, the silica concentration in the effluent will increase proportional to the silica concentration on the resin.
- As the pH increases, so does the silica concentration in the effluent.
- To maintain a low silica concentration as pH (hydroxide concentration) is increased, less silica can be permitted on the resin.
- Salem, E., “A Study of the Chemical and Physical Characteristics of Ion Exchange Media Used in Trace Contaminant Removal,” American Power Conference, Chicago, IL, April 1969.
- Emmett, J. R. and Grainger, P.M., “Ion Exchange Mechanism in Condensate Polishing,” International Water Conference, Pittsburgh, PA, October 1979.
- Darji, J. D. and McGilbra, H. F., “Ion Exchange Equilibrium – A Key to Condensate Polisher Performance,” American Power Conference, Chicago, IL, April 1980.
- Venderbosch, H. W., Snel, A., Overman, L. J. and Kema, N. V., “Concerning the Capacity of Ion Exchange Resins When Removing Trace Impurities from Water,” VGB Feedwater Conference 1971.
- Connelley, E. J., “New Ion Exchanger Method for Extended Run Operation of Utility Condensate Demineralzers,” Industrial Water Engineering, March/April 1979.